
Abstract
Osteosarcoma is the most common malignant bone tumor in adolescents and young adults. Most osteosarcomas are sporadic but the risk of osteosarcoma is also increased by germline variants in TP53, RB1 and RECQL4 genes. ATRX germline variations are responsible for the rare genetic disorder X-linked alpha-thalassemia mental retardation (ATR-X) syndrome characterized by severe developmental delay and alpha-thalassemia but no obvious increased risk of cancer. Here we report two children with ATR-X syndrome who developed osteosarcoma. Notably, one of the children developed two osteosarcomas separated by 10 years. Those two cases raise the possibility that ATRX germline variant could be associated with an increased risk of osteosarcoma.
Similar content being viewed by others


Introduction
Osteosarcoma is the most common primary sarcoma of the skeleton and occurs mostly in adolescents and young adults. Although most cases of osteosarcomas are sporadic, the risk of osteosarcoma is increased in patients with various germline gene variants including RB1 [1], RECQL4 [2] and TP53 [3, 4]. These osteosarcoma-predisposing genes have provided some insight into the roles of key pathways, such as genetic instability and cell cycle deregulation, during osteosarcoma oncogenesis.
Germline variants in ATRX gene are responsible for a very rare genetic disease called X-linked alpha-thalassemia mental retardation (ATR-X) syndrome [5]. ATR-X syndrome is inherited in an X-linked manner and affects almost only males. ATR-X syndrome is characterized by severe developmental delay, characteristic facial dysmorphism, urogenital abnormalities, as well as mild-to-moderate microcytic anemia related to alpha-thalassemia.
Until the recent report of three patients with ATRX germline variant who all developed osteosarcomas [6, 7], ATR-X syndrome was not known to be associated with an increased risk of cancer. Here we present the case of two additional children with ATRX germline variants and clinical ATR-X syndrome who developed osteosarcomas. Notably one of the children developed two osteosarcomas 10 years apart.
Materials and methods
ATRX analysis
The ATRX gene was analyzed as described before [8]. All primers sequences are available upon request.
CGH (Comparative Genomic Hybdridisation) array
The array used for copy number profiling of the tumors are custom 180K CGH/LOH (Loss of Heterozygosity) from Agilent. Labeling and hybridization were performed as recommended in the manufacturer’s protocol.
Results
Case reports
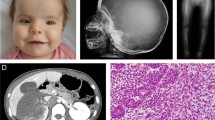
The first patient is a boy who suffered early severe psychomotor retardation, important feeding problems, bilateral cryptorchidism and a facial dysmorphism that strongly suggested ATR-X syndrome. Germline ATRX sequencing revealed the nonsense variant c.7156C > T p.(Arg2386*) (NM_000489.3) (LOVD variant id #0000223794; https://databases.lovd.nl/shared/genes/ATRX) and confirmed the ATR-X syndrome. It is noteworthy that the variant was inherited from his mother and that the first cousin of our patient also inherited the same variant from his mother and also suffered from ATR-X syndrome. Our patient also has one younger brother and one younger sister who are both healthy. The family is of European origin and there was no parental consanguinity. Our patient presented an osteogenic sarcoma of the upper end of the left tibia at the age of 9 years. The tumor was amputated without any additional treatment. Lung metastases appeared 1 year later and were treated by chemotherapy and thoracotomy. Patient was in complete response at the end of the treatment. Ten years later, at the age of 20, a second osteosarcoma occurred in the upper end of the left humerus (Fig. 1c). Compared with the first osteosarcoma, this second tumor harbored very different histological features (Figs. 1a, b, d, e). Chemotherapy and complete resection were conducted. One year after the end of treatment, the patient is still in remission. An array-CGH was performed to investigate somatic copy number alterations (CNAs) patterns in these two separate tumors (Fig. 2). Some CNAs are present in both tumors but affect mostly whole chromosome arms: gain of chromosome 5p and losses of chromosomes 5q, 6q, 8p, 10q and 13. Conversely, some CNAs are only observed on the first or the second tumor including loss of chromosome 3p together with a gain of the terminal region of 14q specifically detected in the first tumor and loss on chromosome 2q, as well as gain of chromosome 8q specifically observed in the second tumor. We also observed shorter CNA specifically observed in the second osteosarcoma including several gains on chromosome 4 and 7p. An amplification encompassing the KIT oncogene (on 4q12) was also specifically observed in the second osteosarcoma.

Hematoxylin and eosin staining of the first osteosarcoma showed predominant sclerosing areas with tumoral cords and lobules embedded in a network of dense collagen strands a. Few areas were made of more cellular proliferation intermingled with calcified osteoid (arrow, b). MRI of the left arm performed 10 years later showed a second osteosarcoma of the upper part of the humerus infiltrating the nearby soft tissue c. Compared with the first osteosarcoma, this second tumor harbored very different histological features that correspond to a conventional high-grade osteosarcoma, without sclerosing areas, with more frequent atypical nuclei and cells, and with a higher cellular density d, e

Array-CGH performed on the primary and the second osteosarcomas of patient 1 identified copy number alterations (CNAs) specifically acquired in those tumors: loss of chromosome 3p and of a telomeric part of 14q in the first tumor; loss on chromosome 2q, gain of chromosome 8q, and several shorter CNA gains on chromosome 4 (including one amplification encompassing the KIT oncogene) and 7p in the second tumor
The second patient is a boy who suffered from severe developmental delay, behavioral disorder, microcephaly, bilateral cryptorchidism and characteristic facial dysmorphism that also strongly suggested ATR-X syndrome. Germline ATRX sequencing revealed a missense variant c.6532C > T p.(Arg2178Trp) (NM_000489.3) (LOVD variant id #0000223795; https://databases.lovd.nl/shared/genes/ATRX) in the helicase domain of the protein. This variation has never been reported in population databases such as ESP, 1000g, ExAC and gnomAD. The amino-acid affected is highly conserved and the distance between the tryptophan that results from this substitution and the arginine original amino acid is high (Grantham score = 101 [0–215]). In silico predictions (SIFT, PolyPhen and Mutation taster) hence revealed a strongly deleterious effect of this variant, which could therefore be considered to likely affects the function of ATRX. Furthermore, this variant has already been described in patient with ATR-X syndrome [8]. The patient developed an osteosarcoma of the left femoral neck with bilateral pulmonary nodules at the age of 4 years, a particularly young age for an osteosarcoma that rarely occurs before 10 years [9]. He was treated by chemotherapy and surgery. Notably, a significant toxicity was observed from chemotherapy (high-dose methotrexate and etoposide-ifosfamide). The tumor relapsed as a metastatic disease in the right lung 18 months after the end of the treatment and rapidly caused the death of the patient.
Discussion
As ATR-X syndrome was not known to be associated with any cancer risk, a coincidence for our two cases must be ruled out. Considering our patient who has developed two metachronous osteosarcomas, it is worth mentioning that (i) osteosarcoma metastasis typically occurs in the lung and rarely in bones, (ii) when they occurred, bone relapses are most often located in the same affected bone, (iii) time lapse of 10 years between the two osteosarcomas is substantially longer than the usual time lapse for relapse: a retrospective follow-up analysis of 101 patients with osteosarcoma recently revealed that most relapses occurred within the first 2 years, whereas only two patients experienced a relapse between years 2 and 5, and none after 5 years [10] and (iv) the histological aspects of these two osteosarcomas are significantly different (Figs. 1a, b, d, e). Taken together, these data indicate that this second tumor harbored clinico-bio-radiological features of a primary tumor and would represent a second independent osteosarcoma, which is highly suggestive of a true cancer predisposition. It should also be emphasized that the ages of onset in both patients are very early for osteosarcomas (i.e., 9 years for cases 1 and 4 years for case 2) compared with the other reported cases of those tumors that usually rather occurs after 10 years [9]. Although RB1, RECQL4 and TP53 alterations have not been excluded in both patients, no personal or familial history suggested such germline events. Altogether those two cases emphasize the possibility that those germline variants had promoted the formation of osteosarcomas in our patients. Furthermore, recent reports from two different teams also described three additional cases of patients with ATR-X syndrome who developed osteosarcomas [6, 7]. The rarity of ATRX germline variants with only a limited number of cases described so far could explain why this association has not been described sooner. In such a rare disorder, a low penetrance for the tumor phenotype may preclude to assess the actual risk of developing cancer.
Interestingly, germline ATRX variants causing the ATR-X syndrome are predominantly missense variants frequently clustered in the ADD (ATRX-DNMT3-DNMT3L) or the helicase protein domains [11]. Conversely, somatic alterations are rather truncating variants such as frameshift and nonsense variations in malignant gliomas [12] and frameshift or nonsense variations and intragenic deletions in osteosarcomas [13]. Remarkably, our first patient, who developed two distant osteosarcomas, has a nonsense germline variant of ATRX. It should also be noticed that the exact same variant was reported in two brothers with ATR-X syndrome and osteosarcomas recently described [6]. This germline truncating variant, compared with the less damaging missense variants, could hence represent a predisposing factor to develop osteosarcomas. However, such a genotype–phenotype correlation remains speculative and it has been demonstrated that some missense variants affect the ATRX function by disrupting the subcellular localization of the protein and have the same functional significance as truncating variations [14]. However, a peculiar attention should be given to the potential other ATR-X syndrome patients who carry the same germline variant.
ATRX germline variations are usually inherited from heterozygous carrier mothers rather than occurring de novo [11]. Heterozygous female carriers have a highly skewed pattern of X-inactivation resulting in preferential inactivation of the variant-bearing X chromosome [8, 11]. Almost all female carriers have normal appearance and intellect, although some show subtle signs of alpha-thalassemia. No tumor predisposition has been described in these women but it would be interesting to determine if asymptomatic women who developed sporadic osteosarcomas carry germline heterozygous variation in ATRX. Interestingly, neither the cousin of our first patient, who is now 6 years old, or his mother or aunt who all carry the same c.7156C > T p.(Arg2386*) variant in ATRX do not suffer from any kind of cancer for now.
It is noteworthy that ATRX represents one of the most frequently somatically altered genes in osteosarcoma together with TP53 and RB1 [13]. Thus, just as TP53 and RB1 germline variations contribute to osteosarcoma predisposition by inducing genomic instability and cell cycle escape, respectively, ATRX germline variants could also lead to osteosarcoma formation through alternative lengthening of telomeres [15, 16] and/or replication deficiency [17]. In accordance with its role in tumor formation, ATRX is mutated at high percentage in various types of tumor in addition to osteosarcomas including high-grade glioma [12], endometrial carcinoma [18], cutaneous squamous cell carcinoma [19], small cell lung cancer [20] and late-onset neuroblastoma [21]. Thus, the involvement of ATRX germline variations as predisposing to osteosarcoma would be consistent with their somatic alteration frequency in sporadic osteosarcomas.
Altogether, we raise strong arguments in favor of ATR-X syndrome being responsible for a predisposition to osteosarcoma and this information should now be considered for genetic counseling in this rare disorder.
References
Kleinerman RA, Tucker MA, Tarone RE, et al. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol. 2005;23:2272–9.
Hicks MJ, Roth JR, Kozinetz CA, Wang LL. Clinicopathologic features of osteosarcoma in patients with Rothmund-Thomson syndrome. J Clin Oncol. 2007;25:370–5.
McIntyre JF, Smith-Sorensen B, Friend SH, et al. Germline mutations of the p53 tumor suppressor gene in children with osteosarcoma. J Clin Oncol. 1994;12:925–30.
Gokgoz N, Wunder JS, Mousses S, Eskandarian S, Bell RS, Andrulis IL. Comparison of p53 mutations in patients with localized osteosarcoma and metastatic osteosarcoma. Cancer. 2001;92:2181–9.
Gibbons RJ, Wilkie AO, Weatherall DJ, Higgs DR. A newly defined X linked mental retardation syndrome associated with alpha thalassaemia. J Med Genet. 1991;28:729–33.
Ji J, Quindipan C, Parham D et al. Inherited germline ATRX mutation in two brothers with ATR-X syndrome and osteosarcoma. Am J Med Genet A. 2017. https://doi.org/10.1002/ajmg.a.38184
Smolle MA, Heitzer E, Geigl JB et al. A novel mutation in ATRX associated with intellectual disability, syndromic features, and osteosarcoma. Pediatr Blood Cancer. 2017;64:e26522. https://doi.org/10.1002/pbc.26522.
Badens C, Lacoste C, Philip N, et al. Mutations in PHD-like domain of the ATRX gene correlate with severe psychomotor impairment and severe urogenital abnormalities in patients with ATRX syndrome. Clin Genet. 2006;70:57–62.
Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3–13.
Rothermundt C, Seddon BM, Dileo P, et al. Follow-up practices for high-grade extremity Osteosarcoma. BMC Cancer. 2016;16:301.
Gibbons RJ, Wada T, Fisher CA, et al. Mutations in the chromatin-associated protein ATRX. Hum Mutat. 2008;29:796–802.
Cancer Genome Atlas Research Network. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372:2481–98.
Chen X, Bahrami A, Pappo A, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7:104–12.
Cardoso C, Lutz Y, Mignon C, et al. ATR-X mutations cause impaired nuclear location and altered DNA binding properties of the XNP/ATR-X protein. J Med Genet. 2000;37:746–51.
Picketts DJ, Higgs DR, Bachoo S, Blake DJ, Quarrell OW, Gibbons RJ. ATRX encodes a novel member of the SNF2 family of proteins: mutations point to a common mechanism underlying the ATR-X syndrome. Hum Mol Genet. 1996;5:1899–907.
Heaphy CM, de Wilde RF, Jiao Y, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425.
Clynes D, Gibbons RJ. ATRX and the replication of structured DNA. Curr Opin Genet Dev. 2013;23:289–94.
Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73.
Li YY, Hanna GJ, Laga AC, Haddad RI, Lorch JH, Hammerman PS. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin Cancer Res. 2015;21:1447–56.
George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53.
Pugh TJ, Morozova O, Attiyeh EF, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–84.
Acknowledgements
The authors thank the “Tumorothèque Necker-Enfants Malades”.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Masliah-Planchon, J., Lévy, D., Héron, D. et al. Does ATRX germline variation predispose to osteosarcoma? Three additional cases of osteosarcoma in two ATR-X syndrome patients. Eur J Hum Genet 26, 1217–1221 (2018). https://doi.org/10.1038/s41431-018-0147-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0147-x
This article is cited by
-
Germline mutations of homologous recombination genes and clinical outcomes in pancreatic cancer: a multicenter study in Taiwan
Journal of Biomedical Science (2024)
-
Targeted therapy for osteosarcoma: a review
Journal of Cancer Research and Clinical Oncology (2023)
-
Synchronous lung and multiple soft tissue metastases developed from osteosarcoma of tibia: a rare case report and genetic profile analysis
BMC Musculoskeletal Disorders (2022)
-
ATR-X syndrome: genetics, clinical spectrum, and management
Human Genetics (2021)
-
G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma
Nature Communications (2019)
